Calvin Chi
calvin.chi at berkeley dot edu
I am a senior applied scientist in a new AI team formed in 2025 at Amazon, focused on training foundational models and building LLM agents in the space of advertising. I am currently the lead scientist in developing an external-facing conversational agent for one of our core products. Previously, I was in Amazon Advertising's Demand Side Platform team, where I helped launch the team's first deep learning conversion prediction and pre-ranking models. I also led the design, research, and launch of a bandit algorithm in the bidding process as the first exploration mechanism in the system.
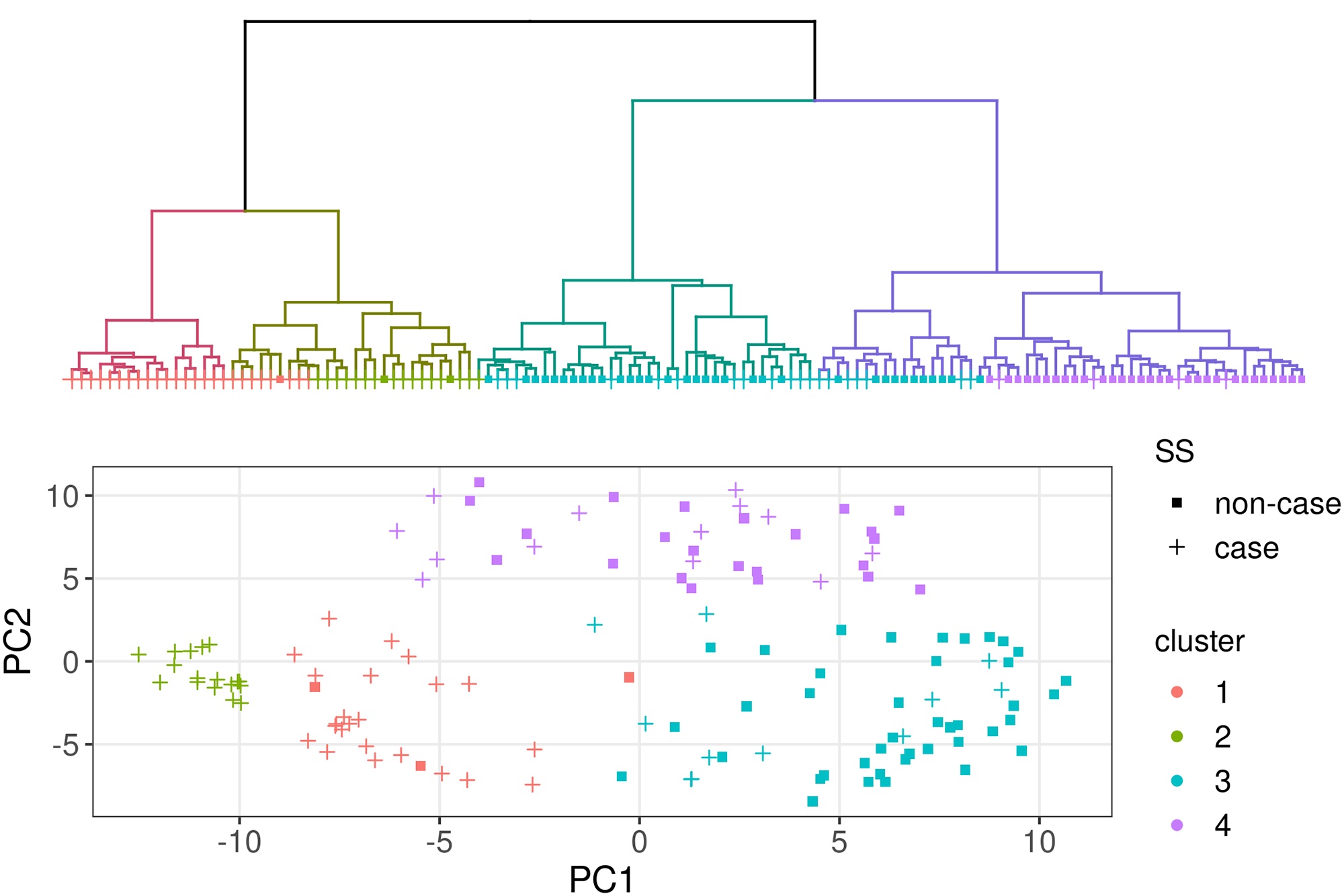
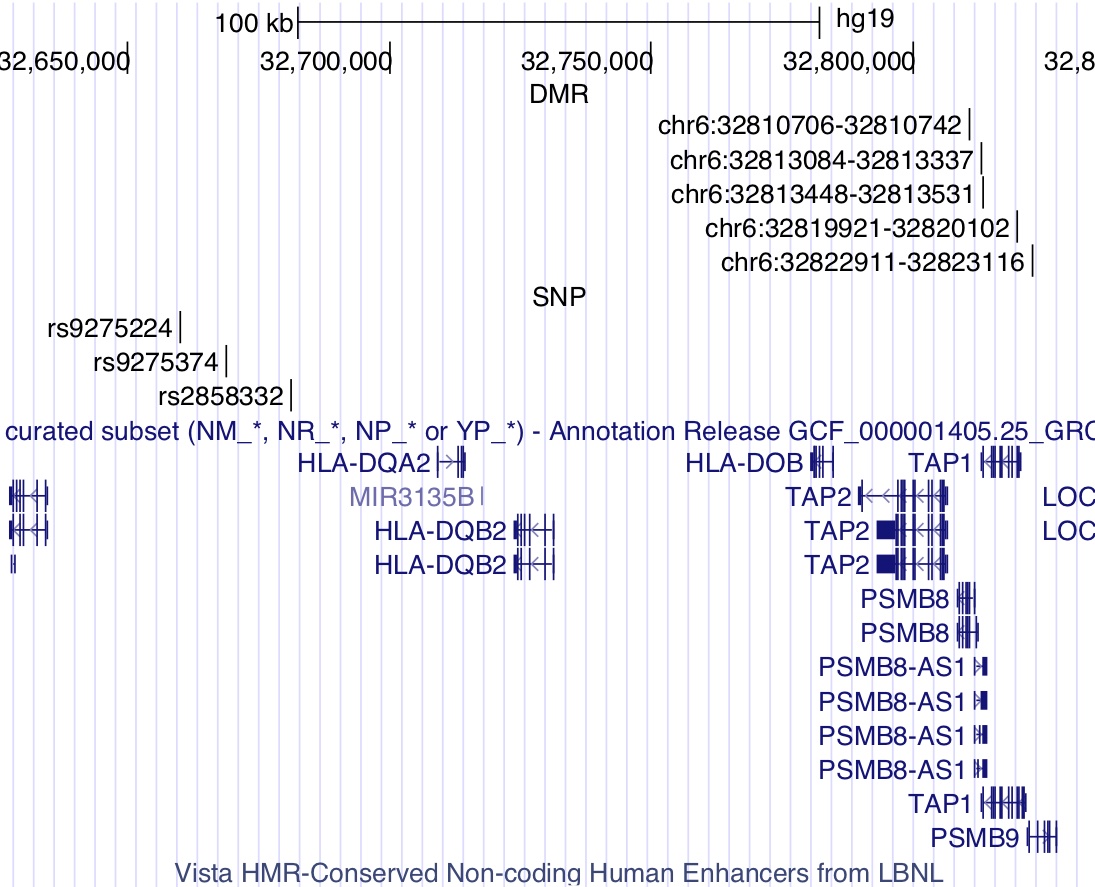
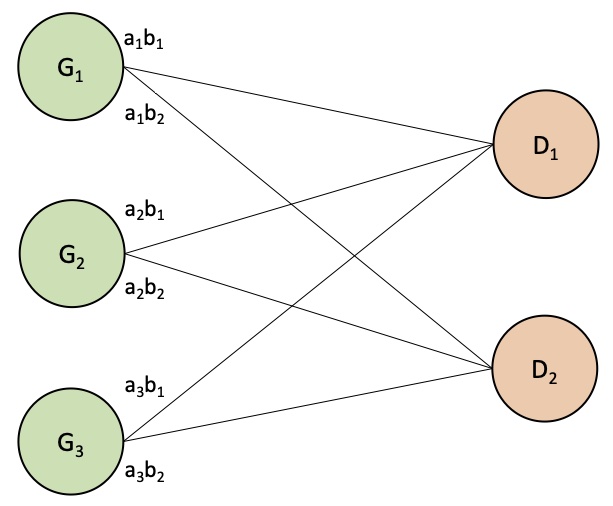
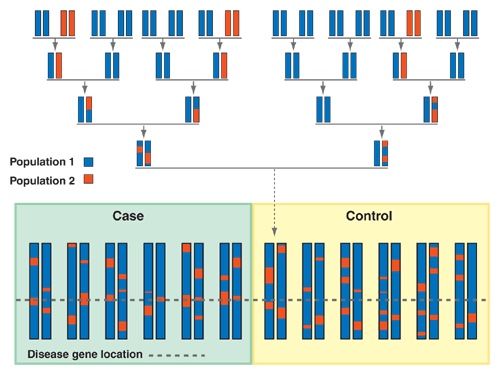
I completed my Ph.D. in Computational Biology at UC Berkeley in 2020, advised by Haiyan Huang and Lisa Barcellos, and supported by the NSF Graduate Research Fellowship. My graduate research was at the intersection of applied machine learning and epidemiology & pharmacogenomics, which is summarized by this dissertation.
Interest wise, I love both the theory behind ML, optimization, reinforcement learning, and the practical building of AI systems. I am currently writing a book about LLM agents.
"Nature cannot be fooled." - Richard Feynman